






Projects Summary
We investigate two broad and related areas:
- The mechanisms and physiological consequences of alternative splicing transitions during mammalian postnatal development
- The disruption of this regulatory network that is the pathogenic mechanism of the repeat expansion disease myotonic dystrophy type 1 (DM1)
There are several potential research projects at any given time; examples are below and new ideas are welcome. Contact Dr. Cooper to explore options.
Mechanism of Myotonic Dystrophy Pathogenesis
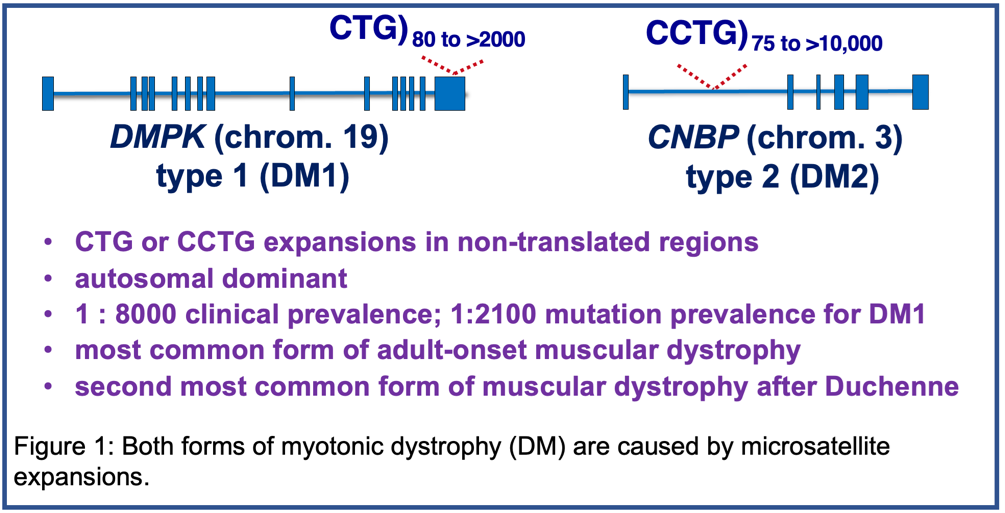
Myotonic Dystrophy (DM)
Myotonic dystrophy (DM) is the second most common cause of muscular dystrophy and the most common cause of adult onset muscular dystrophy with a clinical prevalence of 1:8000. There are two forms of DM that are both caused by microsatellite expansions. DM type 1 (DM1) is caused by a CTG expansion in the 3' untranslated region of the DMPK gene on chromosome 19 and type 2 (DM2) is caused by a CCTG expansion in intron 1 of the CNBP gene on chromosome 3 (Figure 1). Our focus is DM1, the more severe form, although the pathogenic mechanism is similar for both DM1 and DM2. DM1 is autosomal dominant and the disease severely affects multiple tissues including skeletal muscle, heart, central nervous system and gastrointestinal (GI) system. The mutation prevalence of DM1 has been shown to be 1:2100. For additional information about DM including resources for affected families, see “Myotonic Dystrophy Links.”
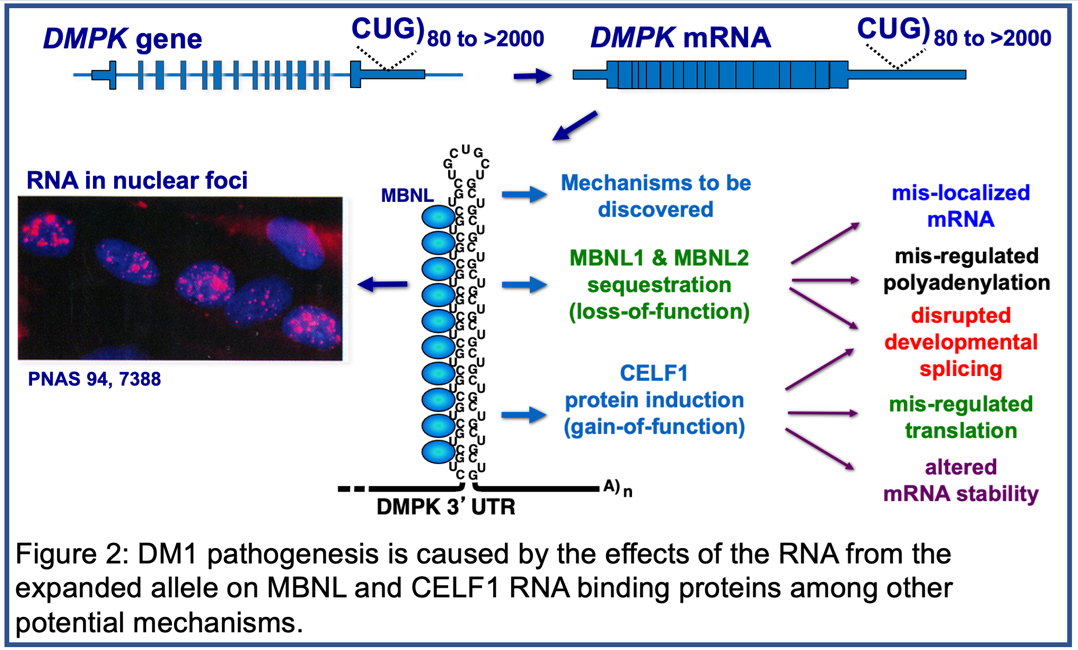
DM1 Pathogenic Mechanisms: Disruption of Developmentally Regulated RNA processing
Pathogenic DMPK alleles contain from 50 to thousands of CTG repeats in the 3’ untranslated region of the gene compared to 5-37 CTG repeats in non-pathogenic alleles. The mRNA expressed from the expanded allele, containing long tracts of CUG repeats (CUGexp RNA), is toxic due to disruption of the functions of at least two families of RNA binding proteins: MBNL and CELF. CUGexp RNA forms based-paired structures that bind and sequester MBNL protein resulting in a loss of function while CELF protein expression is up regulated (Figure 2). Members of these two families regulate multiple aspects of post-transcriptional gene expression during heart and skeletal muscle postnatal development including alternative splicing, selection of polyadenylation sites, translation, mRNA stability, and mRNA localization.
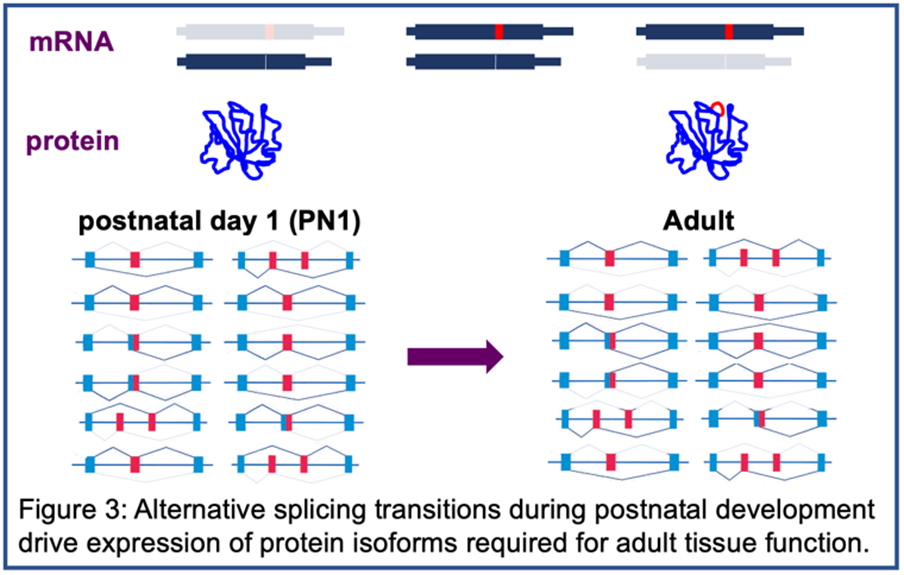
The best-characterized effect of CUGexp RNA is disrupted splicing regulated by MBNL and/or CELF proteins leading to a failure to transition to adult splicing patterns causing the expression of fetal protein isoforms in adult tissues. The inability of fetal isoforms to fulfill the functions required in adult tissues is the molecular basis for disease features such as myotonia however the mechanisms causing muscle wasting, cardiac arrhythmias and GI dysfunction are unknown and are major areas of investigation in our lab.
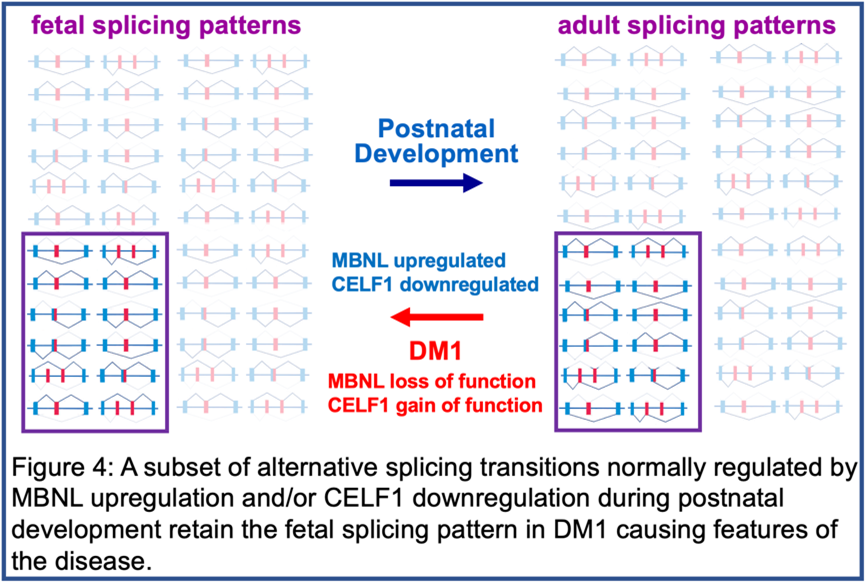
It was the study of DM1 that revealed the network of coordinated alternative splicing transitions that occur after birth as tissues remodel from fetal to adult function (Figures 3 and 4). The normal function of this network is the second area of investigation in the lab, described below.
Representative papers: PNAS 105, 2646; J Clin Invest 119, 3797; PNAS 110, 13570; Cell Reports 6, 336, PNAS 109, 4221, ACS Chem Biol 12, 2503; Human Mol. Genet. 27, 2789; J Am Heart Assoc 7, e010393; J Clin Invest Insight 6:e143465; Nucleic Acids Research 2023 10.1093/nar/gkac1219 and check out the most recent publications.
DM1 Project Areas
We developed tissue-specific tetracycline-inducible DM1 mouse models to express human DMPK RNA containing 960 CUG repeats that reproduce several phenotypic and molecular features of DM1 in heart and skeletal muscle (Human Mol. Genet. 27, 2789; J Clin Invest Insight 6:e143465). We also use immortalized myoblast cell culture models of the disease. Specific projects include:
- Identify the molecular basis for skeletal muscle wasting using the DM1 CUGexp RNA mouse model, immortalized human DM1 muscle cultures and DM1 patient tissue samples.
- Identify the molecular basis for cardiac arrhythmias using the DM1 CUGexp RNA mouse model and patient tissue samples.
- Conditional knock out mice for Mbnl1 and Mbnl2 are being used to determine the mechanisms of skeletal muscle wasting, cardiac features and disrupted GI smooth muscle function in DM1.
- Identify proteins associated with foci and non-foci CUGexp RNA using proximity labeling to define altered signaling pathways, disease modifiers and potential therapeutic targets.
- Determine the mechanisms of pathogenic effects of CUGexp RNA in addition to MBNL loss of function including upregulation of CELF1, other RNA binding proteins and altered signaling.
- Immortalized DM1 myoblast cultures will be used for screens to identify genetic modifiers of hallmark splicing changes to be applied to the mouse models and validated in patient tissue samples.
- The DM1 CUGexp RNA mouse model is being used to test therapeutic approaches including with industrial partners.
It is an exciting time in the DM field in that a large number of pharmaceutical companies are investing in therapeutic approaches including clinical trials. Our mouse models are being used by several companies for preclinical testing including in our collaborations in Sponsored Research Agreements. Combinatorial therapeutic approaches are likely to be required given the multiple tissues affected in DM1. Our mechanistic studies are designed with the goal of identifying robust therapeutic targets.
Mechanisms and Consequences of Coordinated Splicing Regulation
We are using three experimental systems to identify previously unknown roles for coordinated alternative splicing and additional post-transcriptional regulatory mechanisms during periods of physiological change: postnatal heart development, postnatal skeletal muscle development and differentiation of cultured skeletal muscle myoblasts. Cell culture and mouse development provide complementary experimental strategies to develop approaches to test hypotheses relevant to tissue function.
Postnatal development is a period of dynamic transcriptional and post-transcriptional gene regulation driving tissue remodeling from fetal to adult physiology. For example, within two weeks after birth the cardiomyocytes of mouse heart stop proliferating and heart growth occurs by cell hypertrophy, cardiomyocytes transition from glycolytic to oxidative metabolism to match the increased need for energy by a rapidly growing body and the T-tubules and the sarcoplasmic reticulum form to manage Ca+2 handling establishing excitation contraction coupling to synchronize contraction for productive blood flow. There is fascinating biology during postnatal development that has not been explored at the molecular level and our goal is to identify specific molecular mechanisms determinative for critical aspects of adult tissue function.
We used the Genomic and RNA Profiling Core at Baylor College of Medicine to perform RNA-seq during mouse heart and skeletal muscle postnatal development and during C2C12 differentiation and identified extensive transitions in gene expression, alternative splicing, alternative polyadenylation, and alternative promoter usage. The postnatal splicing transitions from fetal to adult isoforms are complete within the first four weeks after birth. In heart, up to 40 percent are conserved between murine and avian development strongly suggesting that the resulting protein isoform transitions are functionally important. In heart and skeletal muscle postnatal development and muscle differentiation in vitro, most genes that produce protein isoform transitions via alternative splicing do not show substantial changes in mRNA levels indicating that alternative splicing produces protein isoform transitions often without a substantial change in gene total output (Nature Comm. 5, 3603; eLife 6:e27192, Cell Reports 24, 197).
Representative papers: PNAS 105, 20333; Genes Dev 24, 653; Nature Comm. 5, 3603; Molecular Cell, 55, 592; eLife 6:e27192, Cell Reports 24, 197; J Am Heart Assoc 7, e010393 and check out the most recent publications.
Developmentally Regulated Splicing Project Areas
General questions driving projects include: What are the dominant regulators of the multiple coordinated splicing networks during postnatal development? What mechanisms are used to modify the activities of the splicing regulators during development (protein abundance, nuclear:cytoplasmic distribution, post-translational modifications)? What signaling pathways are activated to modulate the activities of the regulators? What are the functional consequences of the protein isoform transitions that occur during postnatal development?
- We are using CRISPR/Cas9 to generate mice with genomic deletions of alternative exons that show conserved developmental regulation to force expression of endogenous fetal isoforms through development and in adults. We identified several genes for which failure to express the adult isoform produces a defect in heart and/or skeletal muscle function demonstrating significance to function in vivo. Individual genes provide separate projects that cover different biological processes. This set of projects provides a relatively rapid and transformative approach to determine the functions of postnatal protein isoform transitions in vivo. Many of these splicing events are altered in DM1 and therefore simultaneously investigate mechanisms contributing to postnatal tissue remodeling and DM1 pathogenesis.
- Conditional and cell specific Mbnl knock out or inducible transgenic expression of CELF1 in adult mouse tissues allows identification of early changes responsible for the adult-onset phenotypes. The analysis identifies MBNL and CELF1 biological functions during postnatal development and also provide models to investigate mechanisms of DM1 pathogenesis.
- We developed a splicing reporter that expresses different fluorescent proteins for different splicing patterns to identify cell subpopulations within cultures and mouse tissues that differ in splicing regulation. We will use the reporter in different projects to identify genes required for regulating splicing transitions and to perform screens for modulators of misregulated splicing in DM1.